Published
Author Henry Rzepa

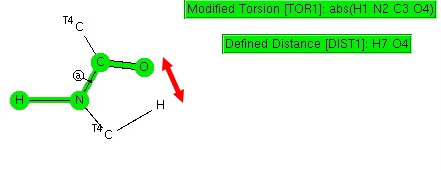
I mentioned in the last post that one can try to predict the outcome of electrophilic aromatic substitution by approximating the properties of the transition state from those of either the reactant or the (presumed Wheland) intermediate by invoking Hammond’s postulate[cite]10.1021/ja01607a027[/cite]. A third option is readily available nowadays; calculate the transition state directly. Here are the results of exploring this third variation.