
Published
Author Henry Rzepa

The mechanism of the reaction of alkenes known as ozonolysis was first set out in its modern form by Criegee. The crucial steps, (a), (b) and (d), are all pericyclic cycloaddition/eliminations.
The mechanism of the reaction of alkenes known as ozonolysis was first set out in its modern form by Criegee. The crucial steps, (a), (b) and (d), are all pericyclic cycloaddition/eliminations.

The Baeyer-Villiger rearrangement was named after its discoverers, who in 1899 described the transformation of menthone into the corresponding lactone using Caro’s acid (peroxysulfuric acid). The mechanism is described in all text books of organic chemistry as involving an alkyl migration.

During the 1960s, a holy grail of synthetic chemists was to devise an efficient route to steroids. R. B. Woodward was one the chemists who undertook this challenge, starting from compounds known as dienones ( e.g. 1 ) and their mysterious conversion to phenols ( e.g. 2 or 3 ) under acidic conditions.

Astronomers who discover an asteroid get to name it, mathematicians have theorems named after them. Synthetic chemists get to name molecules (Hector’s base and Meldrum’s acid spring to mind) and reactions between them. What do computational chemists get to name? Transition states! One of the most famous of recent years is the Houk-List.
Chemists love a mystery as much as anyone. And gaps in patterns can be mysterious. Mendeleev’s period table had famous gaps which led to new discovery. And so from the 1890s onwards, chemists searched for the perbromate anion, BrO 4 – . It represented a gap between perchlorate and periodate, both of which had long been known. As the failure to turn up perbromate persisted, the riddle deepened.

I mentioned in my last post an unjustly neglected paper from that golden age of 1951-1953 by Kirkwood and co. They had shown that Fischer’s famous guess for the absolute configurations of organic chiral molecules was correct. The two molecules used to infer this are shown below.

I wrote in an earlier post how Pauling’s Nobel prize-winning suggestion in February 1951 of a (left-handed) α-helical structure for proteins[cite]10.1073/pnas.37.4.205[/cite] was based on the wrong absolute configuration of the amino acids (hence his helix should really have been the right-handed enantiomer). This was most famously established a few months later by Bijvoet’s[cite]10.1038/168271a0[/cite] definitive crystallographic
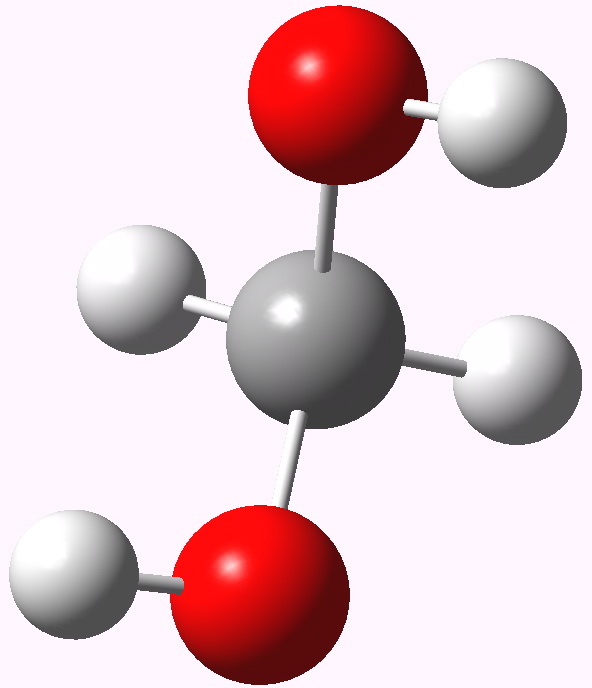
In my previous post I speculated why bis (trifluoromethyl) ketone tends to fully form a hydrate when dissolved in water, but acetone does not. Here I turn to asking why formaldehyde is also 80% converted to methanediol in water? Could it be that again, the diol is somehow preferentially stabilised compared to the carbonyl precursor and if so, why? Methanediol. The lowest energy geometry is shown above.

The equilibrium for the hydration of a ketone to form a gem-diol hydrate is known to be highly sensitive to substituents. Consider the two equilibria: For propanone, it lies almost entirely on the left, whereas for the hexafluoro derivative it is almost entirely on the right. The standard answer to this is that electron-withdrawing substituents destabilize the carbonyl compound more than the hydrate.
Sometimes, as a break from describing chemistry, I take to describing the (chemical/scientific) creations behind the (WordPress) blog system. It is fascinating how there do seem increasing signs of convergence between the blog post and the journal article. Perhaps prompted by transclusion of tools such as Jmol and LaTex into Wikis and blogs, I list the following interesting developments in both genres.